retinochoroidite de type birdshot
Présentation de la rétinochoroïdite de type birdshot par l’Association Horus birdshot -

La rétinochoroïdite de type birdshot est maladie orpheline auto-immune (Les maladies auto-immunes résultent d'un dysfonctionnement du système immunitaire conduisant ce dernier à s'attaquer aux constituants normaux de l'organisme. C'est par exemple le cas dans le diabète de type 1, la sclérose en plaques ou encore la polyarthrite rhumatoïde.) potentiellement cécitante.
Sa prévalence (En épidémiologie, la prévalence est une mesure de l'état de santé d'une population, dénombrant le nombre de cas de maladies, à un instant donné ou sur une période donnée. Pour une affection donnée, on calcule le taux de prévalence en rapportant ce nombre à la population considérée) est de 0.35 (soit environ 1 cas par 300.000).
Au sein de l'Union européenne, une maladie est dite rare lorsqu'elle affecte moins d'une personne sur 2 000. Ainsi en France, on dit qu'une maladie est rare si elle affecte moins de 30 000 personnes. Les maladies rares concernent 3 à 4 millions de personnes en France et près de 25 millions en Europe.
Sommaire :
· Création de l’association Horus birdshot
· La maladie
· Diagnostic
· Symptomatologie
· Examen clinique
· Évolution
· Complications
· Examens complémentaires
· Diagnostic différentiel
· Les traitements
o 9.1Les médications
o 9.2Les traitements alternatifs
o 9.3La diététique "anti inflammatoires"
· Les examens
· Les solutions actuelles et celles d'un futur proche
· Les Hôpitaux et médecins qui traitent la maladie dans le monde
· Les références
Création de l’association Horus birdshot :
L’association Horus birdshot est une association loi 1901 à but non lucratif, dédié à la rétinochoroïdite de type birdshot, maladie auto-immune potentiellement cécitante. C’est une uvéite postérieure non infectieuse.
Elle a été créée à Reims, le 11 décembre 2010 (inscription à la préfecture de Reims
Identification R.N.A. : W513001893 - journal officiel du 11/12/2010 - annonce n°937 - No de parution : 20100050) par Sylvain MIGNON. (lien : http://www.horus-birdshot.fr/mon-histoire-mes-traitements.html)
Elle regroupe à ce jour 4000 personnes qui nous suivent sur les réseaux sociaux : Des malades, leurs familles et amis, mais également beaucoup de professionnels de l’ophtalmologie, et ce, dans plus de 60 pays.
La maladie
La choriorétinopathie type Birdshot est une inflammation granulomateuse, l’inflammation granulomateuse est un aspect particulier de la réponse inflammatoire chronique secondaire à la persistance de l'agent étiologique. Les cellules du système des phagocytes mononuclées s'organisent en amas ou en nodules appelés granulomes de la choroïde et de la rétine caractérisée par des lésions choroïdiennes hypo pigmentées multiples de couleur orange-crème dans le fond d'œil, avec une forte association à l'antigène HLA A29. Il s'agit d'une maladie chronique et progressive d'atteinte oculaire. Les patients présentent :
· Une vision trouble
· Des corps flottants
· Une photopsie (Les photopsies sont provoquées non par la lumière, mais par un choc sur le globe oculaire ou l'excitation électrique de la rétine. Le phénomène est souvent associé avec un détachement vitreux postérieur, la migraine, un décollement de rétine ou l'infarctus du lobe occipital.)
· Des scotomes (Un scotome est provoqué par un dysfonctionnement au niveau de la rétine ou du nerf optique, qui génère une tache sombre ou lumineuse dans le champ de vision.)
· Une héméralopie pouvant entraîner la perte de la vision.
L'introduction précoce d'une thérapie immunomudulatrice est importante pour préserver la vision et permettre une rémission1.
La rétinochoroïdite de type birdshot est une maladie inflammatoire oculaire chronique touchant surtout le segment postérieur de l'œil.
La première description de cette maladie est probablement due à Franceschetti et Babel (1949) qui l'ont appelée « choriorétinopathie en taches de bougie ».
Elle est individualisée en 1980 par Ryan et Maumence qui la nomment " Birdshot retinochoroïdopathy " en raison de son aspect caractéristique au fond d'œil montrant de nombreuses petites taches crémeuses disséminées comme " une volée de plombs de chasse ". Cet aspect caractéristique du fond d'œil et la très forte association à l'antigène HLA-A29.2 en font une entité distincte des autres uvéïtes postérieures.
Il s'agit d'une maladie rare (moins de 1,5% des uvéïtes), plus fréquente chez les caucasiens d'origine Nord-Européenne, survenant entre 35 et 70 ans, l'âge moyen étant 50 ans.
Le diagnostic est essentiellement clinique, le typage HLA venant le conforter. Bien qu'il existe des formes bénignes avec une simple perception de corps flottants, la diminution de l'acuité visuelle peut être très sévère. L'atteinte est bilatérale avec présence de taches de couleur crème disséminées au fond d'œil situées au niveau de l'épithélium pigmentaire rétinien ou de la choroïde.
L'évolution est lente avec des périodes d'exacerbation et de rémission expliquant une diminution insidieuse et progressive de la vision. La complication la plus fréquente, menaçant la vision centrale en l'absence de traitement précoce, est l'œdème maculaire cystoïde (Qui ressemble à une vessie. ), environ 60% des cas.
L'étiologie ( Étude des causes des maladies. PAR EXTENSION Ensemble des causes d'une maladie) de la maladie est inconnue mais des mécanismes auto-immuns, dirigés contre des antigènes rétiniens, semblent jouer un rôle dans l'entretien des phénomènes inflammatoires intra-oculaires.
Il n'existe actuellement pas de consensus thérapeutique car aucune étude contrôlée n'a pu être réalisée en raison de la rareté de la maladie.
Bien qu'une relative cortico-résistance ait été d'emblée signalée dans les articles initiaux, le traitement reposait, jusqu'à ces dernières années, soit sur les corticoïdes en injections péri-oculaires ou par voie générale, soit sur la ciclosporine seule ou associée à une faible dose de corticoïdes ou d'un autre immunomodulateur, l'azathioprine.
Fin des années 1990, des résultats intéressants ont été obtenus avec l'administration répétée de TÉGÉLINE® (immunogloguline humaine normale IV). Deux études, l'une prospective (n = 18) et l'autre rétrospective (n = 37), confirment l'excellente tolérance et établissent l'efficacité de TÉGÉLINE® dans le traitement de la rétinochoroïdite de type birdshot. Dans la dernière étude, Tégéline a permis d'améliorer l'acuité visuelle chez 70% des patients étudiés (23/33) et les lésions ongiographiques dans 69% des cas (18/26) ainsi que la diminution ou l'abstention de traitement corticoïde chez plus de la moitié des patients (19/37).
Diagnostic
Le diagnostic de rétinochoroïdite de type birdshot est essentiellement clinique. Le typage HLA peut venir le conforter, l'antigène HLA-A29 étant présent dans 80 à 98% des cas selon les séries.
Symptomatologie
Les patients se plaignent en général d'une perception de corps flottants avec une diminution variable de la vision. Ils décrivent souvent une photophobie, une difficulté dans la distinction des couleurs et une héméralopie (trouble de la vision crépusculaire et nocturne). Ces plaintes visuelles sont fréquemment disproportionnées par rapport à la mesure de l'acuité visuelle centrale, illustrant un dysfonctionnement rétinien diffus.
Examen clinique
La description initiale de Ryan et Maumence comprend 5 critères diagnostiques2 :
1. Un œil blanc et indolore
2. Une inflammation très réduite du segment antérieur de l'œil.
3. Un trouble inflammatoire du vitré sans « œufs de fourmi » ni exsudat en « banquise », différenciant la rétinochoroïdite de type birdshot de la pars planite.
4. Une fuite vasculaire rétinienne, bien visible à l'angiographie à la fluorescéine, entraînant un œdème maculaire cystoïde, souvent associé à un œdème de la papille optique.
5. Des taches discrètes, mais bien individualisées, dépigmentées ou de couleur crème, dispersées dans tout le fond d'œil en arrière de l'équateur du globe.
L'examen clinique montre peu de signes inflammatoires du segment antérieur bien qu'une minime réaction cellulaire de la chambre antérieure puisse parfois se rencontrer. Il n'existe jamais de synéchies irido-cristalliennes. Les patients ne se plaignent d'aucune douleur. L'atteinte est habituellement bilatérale et caractérisée, comme nous l'avons déjà signalé, par la présence de taches de couleur crème disséminées dans tout le fond d'œil au niveau de l'épithélium (Tissu formé de cellules juxtaposées qui recouvre la surface du corps ou qui tapisse l'intérieur de tous les organes creux. ) pigmentaire rétinien ou de la choroïde (Membrane de l'œil, entre la sclérotique et la rétine.) . Les taches sont souvent plus facilement visibles dans l'aire nasale inférieure. Elles peuvent être présentes selon une distribution variée au niveau du pôle postérieur et de la périphérie moyenne.
Les lésions peuvent être soit diffuses, soit asymétriques avec des taches concentrées surtout dans la partie inférieure. De même, elles épargnent souvent la macula.
Certaines lésions périphériques sont étendues et réparties le long des gros vaisseaux choroïdiens. Elles peuvent se présenter comme irradiant à partir de la papille optique.
Les lésions sont habituellement ovales sans hyperpigmentation et leur taille est d’un quart à trois quart du diamètre de la papille optique. Parfois ces lésions deviennent confluentes.
Chez quelques patients, l'évolution est particulière avec l'apparition tardive de taches hypopigmentées, plusieurs années suivant un syndrome d'inflammation intraoculaire chronique avec papillite (Papille optique : lieu d'émergence de la tête du nerf optique dans la rétine) , trouble inflammatoire du vitré et vasculite (Vascularites systémiques : ce groupe de maladies est caractérisé par une inflammation et une nécrose des parois vasculaires) rétinienne. Dans ces formes anciennes d'inflammation uvéale et des vaisseaux rétiniens sans lésions caractéristiques de couleur crème (dont l'apparition est retardée), 'e typage HLA (recherche de l'antigène HLA-A29) est alors nécessaire pour ne pas conduire à une erreur de diagnostic tel celui de vasculite rétinienne idiopathique.
Chez tous les patients, il existe une réaction cellulaire dans l'humeur vitrée. Le trouble inflammatoire du vitré est habituellement diffus avec souvent une prédominance dans la partie postérieure de la cavité vitréenne. Ils sont plus nombreux et importants aux stades précoces de la maladie, associés parfois à des brides vitréennes. Les cellules peuvent se concentrer sur la face postérieure du vitré pour former des agrégats.
Les autres signes cliniques peuvent comprendre un rétrécissement du calibre artériolaire rétinien, une tortuosité vasculaire, une néo vascularisation rétinienne et pré rétinienne, des hémorragies périvasculaires et des membranes épirétiniennes menaçant la macula. Les vasculites au niveau des veinules rétiniennes peuvent se traduire par un engainement fin d'aspect grisâtre différent des engainement épais, floconneux, jaunâtres retrouvés dans d'autres types de vasculites rétiniennes (sarcoïdose par exemple).
La vasculite rétinienne et l'inflammation diffuse du segment postérieur peuvent entraîner un œdème maculaire, localisé ou diffus, évoluant souvent en œdème maculaire cystoïde. À la papillite avec papille optique hyperhémiée peut progressivement succéder une atrophie optique. Chez de nombreux patients, les taches visibles au fond d'œil sont attenantes à la papille optique, donnant un aspect en pétales.
Une neuropathie optique ischémique antérieure aiguë a été décrite chez un patient présentant une rétinochoroïdite de type birdshot.
Les atteintes du nerf optique passent souvent inaperçues et sont sous-estimées ; elles doivent toujours être recherchées par une surveillance systématique du champ visuel.
Dans de rares cas, après de nombreuses années d'évolution, certains patients peuvent se présenter avec un aspect cicatriciel atrophique diffus de tout le fond d'œil périphérique.
Évolution
La rétinochoroïdite de type birdshot est une maladie chronique d'évolution lente caractérisée par des périodes d'exacerbation et de rémission avec diminution progressive de la vision. La perte de la vision est liée à l'œdème maculaire cystoïde ou à une atrophie optique due à une atteinte des fibres nerveuses.
Sans traitement, un tiers à la moitié des patients, selon les études, auront une baisse de l'acuité visuelle inférieure à 5/10. Si aucun traitement n'est administré, les formes évolutives de la rétinochoroïdite de type birdshot conduisent à une altération visuelle sévère dans les premières années (baisse visuelle inférieure à 3/10, amputation du champ visuel, photophobie, héméralopie).
Dans 20% des cas, une forme atténuée peut être observée avec maintien de la vision (7/10 à 10/10), régression spontanée de l'inflammation intra-oculaire et persistance de taches plutôt atrophiques disséminées dans tout le fond d'œil.
Au bout de 7 à 10 années d'évolution, tandis que le processus évolutif de la maladie devient inactif, les lésions au fond d'œil peuvent devenir partiellement pigmentées, au centre plutôt qu'à la périphérie. Aux stades avancés de la maladie, une atrophie optique et une dépigmentation diffuse du fond d'œil sont souvent observées.
Complications
L'œdème maculaire cystoïde chronique est la complication la plus fréquente. Il survient dans plus de 60% des cas et menace la vision centrale. Il nécessite une prise en charge thérapeutique.
Si le traitement est instauré précocement, avant le développement d'une dégénérescence maculaire cystoïde, il peut réduire l'œdème rétinien et restaurer une bonne vision centrale.
La présence de membranes épirétiniennes et une maculopathie de type cellophane sont des complications assez fréquentes, survenant dans presque 10% des cas. Le plissement maculaire peut s'accroître et progresser pour son propre compte comme un phénomène cicatriciel tandis que l'inflammation intra-oculaire régresse sous traitement.
Des membranes néovasculaires sous-rétiniennes peuvent survenir durant l'évolution de la maladie (environ 6% des cas). Chez certains patients, il existe un lien étroit entre une lésion au fond d'œil et le développement de néovaisseaux secondaire à une cicatrice rétinochoroïdienne atrophique. Six mois à cinq ans après le début de lma maladie, ces membranes néovasculaires peuvent se développer au voisinage des lésions rétinochoroïdiennes du fond d'œil.
Des membranes néovasculaires sous-rétiniennes peuvent aussi concerner l'aire juxtapapillaire ou menacer la vision centrale dans la région juxtafovéolaire. Des angiographies à la fluorescéine et au vert d'indocyanine sont essentielles pour déceler les néovaisseaux sous-rétiniens et guider une éventuelle photocoagulation au laser qui peut éviter une perte de la vision centrale.
Une néovascularisation prérétinienne périphérique et prépapillaire est retrouvée dans 7% des cas, même en l'absence de territoire de non perfusion capillaire. Une néovascularisation prérétinienne périphérique peut être due à un phénomène inflammatoire localisé et isolé dans le lit vasculaire rétinien, sans occlusion des capillaires rétiniens.
Cette forme de néovascularisation peut conduire à une hémorragie intravitréenne.
L'atrophie optique représente une séquelle importante d'un phénomène inflammatoire ancien et prolongé mais peut être parfois liée à une neuropathie optique ischémique (L'ischémie est l'arrêt ou l'insuffisance de la circulation sanguine dans une partie du corps ou un organe, qui prive les cellules d'apport d'oxygène et entraîne leur nécrose ) antérieure aiguë. L'incidence des maladies cardiovasculaires (HTA, coronaropathies, infarctus, occlusions veineuses rétiniennes) et de glaucome à angle ouvert semble élevée chez les patients atteints de rétinochoroïdite de type birdshot.
Examens complémentaires
Aucun examen spécifique ne permet d'établir le diagnostic de rétinochoroïdite de type birdshot qui demeure un diagnostic clinique. Seul le typage HLA, pour la recherche de l'antigène HLA-A29 présent dans 80 à 98% des cas, vient conforter le diagnostic. Le sous-typage HLA-A29 n'apporte rien dans le cadre du diagnostic.
Les réponses immunologiques à certains antigènes rétiniens (antigène S rétinien, Interphotoreceptor Retinoïd Binding Protein ou IRBP) peuvent être positives comme dans d'autres uvéïtes postérieures chroniques et ne sont donc pas spécifiques. Les autres examens complémentaires s'inscrivent dans le cadre du diagnostic différentiel afin d'écarter certaines pathologies comme une syphilis (FTA-Abs), une sarcoïdose (enzyme de conversion, lysosyme, scanner…) un lymphome oculo-cérébral (IRM, PL…).
L'OCT est un examen non invasif, permettant d'étudier l'épaisseur de la rétine et d'évaluer avec précision l'existence ou non d'un œdème maculaire cystoïde.
Diagnostic différentiel
Le diagnostic différentiel peut se poser avec un certain nombre de pathologies pouvant présenter des points communs avec la rétinochoroïdite de type birdshot.
Ce sont essentiellement la pars planite, d'autres types de " White Dot Syndromes " tels que le " Multiple Evanescent White Dot Syndrome " ou les choroïdites multifocales (les " White Dot Syndromes " forment un ensemble sémiologique dont fait partie la rétinochoroïdite de type birdshot), la sarcoïdose et d'autres uvéïtes endogènes ou infectieuses.
La pars planite touche des sujets plus jeunes et se caractérise par un trouble inflammatoire bilatéral prédominant plutôt dans le vitré antérieur, sans taches de couleur crème au fond d'œil. Ces patients présentent des modifications de la base du vitré, de la rétine périphérique et de la pars plana qui donnent l'aspect d' "œufs de fourmi, de flocons ou de banquise ".
Le MEWDS (Multiple Evanescent White Dot Syndrome) se rencontre chez des sujets plus jeunes, âgés de 28 ans en moyenne, et se caractérise par une chute brutale de la vision associée à des taches claires au niveau de l'épithélium pigmentaire rétinien. Ce syndrome est habituellement unilatéral, avec un trouble inflammatoire minime du vitré et, dans la plupart des cas, une amélioration spontanée dans les six semaines.
Les patients atteints de choroïdite multifocale sont plus jeunes que ceux souffrant de la maladie de type birdshot. Ils présentent une atteinte unilatérale, un processus inflammatoire plus important et sont moins propices à l'héméralopie ou aux anomalies électrorétinographiques. Les lésions tendent à devenir hyperpigmentées. Elles sont plus petites et mieux délimitées que celles de la rétinochoroïdite de type birdshot.
Le diagnostic de sarcoïdose doit être écarté avant de porter celui de rétinochoroïdite de type birdshot. Les patients atteints de sarcoïdose peuvent développer des granulomes choroïdiens se présentant sous forme de lésions blanches ou jaunâtres qui peuvent être confondues avec celles de la maladie de type birdshot, particulièrement lorsque les autres signes d'uvéïte granulomateuse ne sont pas retrouvés. Cette absence de signes spécifiques est fréquente chez les patients atteints de sarcoïdose avec présence de foyers choroïdiens profonds.
Dans la littérature, certains patients, décrits comme souffrant de la rétinochoroïdite de type birdshot, ont en fait des signes de sarcoïdose systémique. Il est donc important d'éliminer une sarcoïdose, en raison de ses complications systémiques possibles, avant d'affirmer le diagnostic de rétinochoroïdite de type birdshot.
D'autres pathologies peuvent être aussi concernées par le diagnostic différentiel de la rétinochoroïdite de type birdshot : tuberculose, syphilis, histoplasmose, pneumocystose choroïdienne, lymphome non-hodgkinien intra-oculaire primitif, ophtalmis sympathique, syndrome de Vogt-Koyanagi-Harada et épithéliopathie en plaques.
La choriorétinite de type Birdshot est considérée comme faisant partie des affections auto-immunes, ce qui signifie que, pour raison encore souvent inconnue, le système immunitaire se retourne contre un organe déterminé, comme s'il s'agissait d'un tissu ne faisant pas partie du soi. C'est le même genre de mécanisme qui survient lors d'une greffe d'organe non ou peu compatible.
De fait, la plupart des traitements des maladies auto-immunes repose sur le principe d'une immunosuppression, c'est-à-dire qu'on va chercher, dans une certaine mesure, à "désactiver" le système immunitaire afin de stopper le mécanisme de rejet.
Au niveau local se déroulent des mécanismes inflammatoires qui vont, soit insidieusement, soit de façon plus brutale comme dans le cas de l'œdème maculaire cystoïde, être responsables des lésions tissulaires et ainsi des symptômes.
Dans le cas du Birdshot, l'action inflammatoire se porte sur le tissu rétinien, tissu noble permettant la vision. Dans la rétine se situent en effet les cellules nerveuses réceptrices permettant de capter l'information lumineuse, puis de la coder en influx nerveux qui seront ensuite analysés et interprétés par le cortex cérébral.
Les traitements
Les médications
A ce jour, il n'existe pas de traitement3 curatif de la maladie, mais bien des traitements des symptômes inflammatoires :
· Corticothérapie : C'est une maladie inflammatoire qui, pour la grande majorité des cas est traitée par corticothérapie. A la découverte de la maladie il est fréquent qu'il y ait des oedemes maculaires L'œdème maculaire est une affection de la rétine assez fréquente, qui touche sa partie centrale, la macula, et peut conduire à une perte d'acuité visuelle sévère.), il sont traités par le biais d'un bolus de cortisone, une hospitalisation de trois jours, est bien souvent nécessaire. Puis on passe à une longue phase de décroissance par paliers, de corticoïdes, en passant de 1mg/kg/jour jusqu'à 0, si l'inflammation disparait. Dans ce cas c'est ce que l'on appelle un traitement systémique. Il existe des traitements locaux comme les inserts de corticoides intravitréens ou des injections sous tenoniennes.
· Immunosuppresseurs : Beaucoup des traitements immunosuppresseurs ont donc comme cible, ces lymphocytes et visent à diminuer leur nombre et leur activité. Ce sont, à la base, des traitements anti rejets.
· Ciclosporine est un médicament utilisé pour plusieurs types de maladies : pour le traitement préventif du rejet de greffe d'organe lors de transplantation ainsi que pour le traitement de certaines maladies inflammatoires comme la polyarthrite rhumatoïde, le psoriasis ou la dermatite atopique.
· Interférons4 (IFN) sont des protéines (glycoprotéines de la famille des cytokines). Ils sont naturellement produits par les cellules du système immunitaire, mais également par d'autres types cellulaires (cellules dendritiques, mononucléées, épithéliales, etc. ) en fonction des sous types.
· anti TNF Alpha : Les anti-TNFα5 sont des biomédicaments qui bloquent l’action du TNFα. Il s’agit d’anticorps ou de récepteurs dirigés contre cette molécule. Ils ont la capacité de se fixer sur lui et de bloquer l’action du TNFα en excès. Cette action permet une diminution de l’inflammation des tissus.
Les traitements alternatifs
· CBD : le CBD est une molécule extraite du chanvre, aussi appelée cannabis. Mais contrairement à la seconde molécule présente dans la plante appelée Delta-9 TetraHydroCannabinol ou THC, le CBD n’a pas d’effet stupéfiant et n’engendre donc pas de dépendance physique. Le CBD semble avoir des propriétés puissante qui peuvent soulager les troubles inflammatoires au sein du corps humain. Il a montré des résultats prometteurs dans la médiation des réponses immunitaires qui sont associées à diverses maladies telles que l’hypertension, la dépression et la maladie d’Alzheimer. Il a été démontré que le CBD a des propriétés anti-inflammatoires prometteuses avec sa capacité à combattre le stress oxydatif, montrant des effets immunosuppresseurs sur les cellules microgliales et les macrophages, qui sont les cellules qui jouent un rôle essentiel dans l’immunité et l’inflammation. En activant les récepteurs de glycérine, Le CBD a montré des résultats en réduisant la douleur inflammatoire chronique et a montré des effets positifs sur l’inflammation intestinale.La réglementation française permet l’utilisation de certaines variétés de chanvre – dépourvues de propriétés stupéfiantes - à des fins industrielles et commerciales sous trois seules conditions : Le chanvre utilisé doit figurer sur une liste de variétés autorisées ; Seules les graines et les fibres du chanvre peuvent être utilisés, les fleurs étant interdites ; Le chanvre utilisé doit avoir une teneur en THC inférieure à 0,2%.
- Curcuma : Protecteur gastro-intestinal et puissant anti-inflammatoire, le curcuma révèle bien d'autres vertus que son simple usage alimentaire.
La diététique "anti inflammatoires"
Une alimentation anti-inflammatoire, c'est une alimentation sans glucose, avec moins de protéines et de lipides difficiles à digérer. Parmi ces aliments : les fruits (surtout rouges) et les légumes dont les crucifères, particulièrement conseillés. Dans l'ensemble des éléments qui ont une efficacité empirique : lait de chèvres ou de brebis, Pain complet , Riz complet, Pâtes complètes, Sucres non raffinés .
Les examens
· OCT : La tomographie en cohérence optique ou tomographie optique cohérente est une technique d'imagerie médicale bien établie qui utilise une onde lumineuse pour capturer des images tridimensionnelles d'un matériau qui diffuse la lumière, avec une résolution de l'ordre du micromètre
· ERG : L'électrorétinogramme est un examen électrophysiologique. Il est réalisé dans des services spécialisés de neurophysiologie clinique ou en ophtalmologie et permet de diagnostiquer certaines anomalies de la rétine. Il est réalisé grâce à un électrorétinographe lors d'une électrorétinographie
· CHAMP VISUEL : L'examen du champ visuel permet de délimiter cet espace. En pratique, il consiste à explorer les limites de ce champ de vision, en présentant à différents endroits une petite lumière d'intensité variable, tout en demandant à la personne examinée de fixer un point central.
· ACUITE : Les échelles Monoyer sont des tests optométriques, c’est-à-dire servant à déterminer l’acuité visuelle en ophtalmologie, inventés par Ferdinand Monoyer. Il s’agit de planches, les lettres de chaque ligne ont la même taille et la taille croît lorsque l’on descend
· ANGIOGRAPHIES : L'angiographie dure 20 minutes et se déroule comme suit : Application d'une goutte dans les pupilles en vue d'une dilatation. Prise de photos du fond des yeux. Injection d'un colorant fluorescent dans les veines. Deuxième série de photos du fond des yeux.
· TEST HLA A29 : Prise de sang et PCR - Les antigènes HLA sont codés par les gènes d'une région appelée, chez l'Homme, le Complexe Majeur d'Histocompatibilité et il a été trouvé une relation plus ou moins étroite entre les antigènes HLA et des maladies auto-immunes. Les principales recherches d'associations entre HLA de classe I et maladies sont réalisées pour les HLA suivants : 1/HLA A*29 : la choriorétinite birdshot est une forme rare d'uvéite bilatérale postérieure qui provoque une inflammation de la choroïde et de la rétine et peut conduire à une perte complète d'acuité visuelle.
Les solutions actuelles et celles d'un futur proche
· Cellules souches : une cellule souche est une cellule indifférenciée capable, à la fois, de générer des cellules spécialisées par différenciation cellulaire et de se maintenir dans l'organisme par division symétrique ou division asymétrique. Dans les maladies dégénératives de la rétine affectant les photorécepteurs, la transplantation de cellules permettant la restauration de la vision est aujourd’hui envisagée. La dernière décennie a vu des progrès remarquables dans la génération de cellules de rétine à partir de cellules souches pluripotentes humaines avec, en particulier, le développement de systèmes de culture en trois dimensions (3D) permettant la génération d’organoïdes de rétine. Dans cette revue, nous faisons un état des lieux sur les stratégies précliniques menées dans des modèles animaux pour le remplacement des photorécepteurs par des photorécepteurs dérivés de cellules souches et présentons les obstacles importants qui restent à être surmontés.
· Nanotechnologies : Leur technologie est basée sur les nanoparticles sensibles à la lumière de polymère (P3HT NPs) dont la taille est environ 350 nanomètres. Les scientifiques ont développé une prothèse rétinienne liquide artificielle injectable qui travaille à rectifier les photorécepteurs dysfonctionnels. Les chercheurs ont dit que leur travail est une application de nanotechnologie au médicament.
· Rétines artificielles : La rétine est composée de cellules sensibles à la lumière appelées photorécepteurs, dont le but est de transformer les signaux lumineux reçus par l’œil en signaux électriques acheminés vers le cerveau. Ce sont ces cellules qui sont détruites au cours de ces pathologies, ce qui peut mener à la cécité. Le principe d’une rétine artificielle est simple : elle est développée pour se substituer à ces photorécepteurs. Le dispositif est en fait constitué d’implants fixés sous la rétine et composés d’électrodes qui viennent stimuler les neurones rétiniens pour porter les messages au cerveau.
· La cornée artificielle : Le matériau utilisé, 100% synthétique et non dégradable, est placé sous la conjonctive, un site vascularisé qui guérit vigoureusement, pour imiter la structure du tissu conjonctif (un maillage de collagène). Une fois la lentille de l'appareil implantée, offrant une qualité optique équivalente à celle d'une cornée parfaite, il stimule la prolifération cellulaire pour une intégration progressive de l'implant dans l'œil sans déclencher de réponse défavorable du système immunitaire.
· Les chiens guides d'aveugles : Un chien guide d'aveugle est un chien mis à la disposition d'un déficient visuel pour faciliter sa vie quotidienne, et notamment ses déplacements. Ces chiens sont formés dans des écoles spécialisées, et remis à leur futur maître gratuitement.
Les Hôpitaux et médecins qui traitent la maladie dans le monde
Liste des établissements traitement la rétinochoroidite de type birdshot. Ils sont localisés sur une carte accessible par tous : carte "they treat birdshot in the world" - A la date du 16 février 2021, on compte 140 hôpitaux et 180 spécialistes, répartis dans 30 pays. Lien : http://www.horus-birdshot.fr/diseease.html en Français et anglais.
Les références
· https://pubmed.ncbi.nlm.nih.gov/32119303/
· https://pubmed.ncbi.nlm.nih.gov/25434765/
· http://www.scielo.br/scielo.php?script=sci_arttext&pid=S0004-27492015000100016
· https://www.researchgate.net/publication/336121027_Birdshot_Chorioretinopathy
· https://dumas.ccsd.cnrs.fr/dumas-01148444/document
· https://pubmed.ncbi.nlm.nih.gov/22106906/
· https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=FR&Expert=179
· https://www.orpha.net/consor/cgi-bin/SupportGroup_Search.php?lng=FR&data_id=132118&title=Association%20Horus%20Birdshot
· https://www.abbviecare.fr/anti-tnf/tout-savoir-sur-les-anti-tnf-alpha/
· https://www.google.com/maps/d/edit?mid=1QeyDD-MTuE0gPCg5fKTbFeFagUcAyI4O&ll=7.460072759492519%2C0&z=2
· https://drive.google.com/file/d/1-Q8plU0g0Aq0-VDiC-Epd2-AqXTyakMv/view?usp=sharing
· https://institutducerveau-icm.org/fr/actualite/effet-anti-inflammatoire-cannabidiol-compose-non-psychoactif-cannabis/
· https://www.doctissimo.fr/nutrition/lait/choisir-lait-animal
· https://www.eurofins-biomnis.com/services/referentiel-des-examens/page/HLA1/
· https://www.medecinesciences.org/en/articles/medsci/full_html/2020/06/msc200073/msc200073.html
·
· https://www.news-medical.net/health/The-Revolutionary-Development-of-a-Nano-Based-Liquid-Retina-Prosthesis-(French).aspx
· Cellule souche#:~:text=En biologie cellulaire, une cellule,division symétrique ou division asymétrique.
http://www.horus-birdshot.fr/index.html
http://www.horus-birdshot.fr/diseease.html
http://www.horus-birdshot.fr/mon-histoire-mes-traitements.html
http://www.horus-birdshot.fr/birdshot-days.html
↑ Brezin, Antoine P., Directeur de la publication. Guillevin, Loïc, Préface., Uvéites [rapport présenté à la] Société Française d'Ophtalmologie, Elsevier-Masson, dl 2010 (ISBN 978-2-294-71107-7 et 2-294-71107-6, OCLC 690750323, lire en ligne)
↑ Traitements sur le site de l'Association Horus Birdshot
↑ Gresser, Ion., Interferon, Academic Press, 1979- (OCLC 848468056, lire en ligne)
↑ abbviecare.fr
↑ « Phytochemistry of the Genus Curcuma », dans Turmeric, CRC Press, 1er mars 2007 (ISBN 978-0-429-12401-3, lire en ligne), p. 91–126
↑ P. Dieterle, « Electrorétinogramme et sénescence », Ophthalmologica, vol. 143, no 4, 1962, p. 296–299 (ISSN 1423-0267 et 0030-3755, DOI 10.1159/000304244, lire en ligne, consulté le 19 février 2021)

Maladies auto-immunes - INSERM -
La rupture de la tolérance au soi




INFO HORUS -
Les maladies auto-immunes résultent d'un dysfonctionnement du système immunitaire conduisant ce dernier à s’attaquer aux constituants normaux de l’organisme. C’est par exemple le cas dans le diabète de type 1, la sclérose en plaques ou encore la polyarthrite rhumatoïde. Face à ces maladies complexes, les chercheurs développent de nouvelles stratégies thérapeutiques, visant à contrôler le système immunitaire sans pour autant qu’il risque de baisser la garde vis-à-vis des agents pathogènes.
Des dysfonctionnements du système immunitaire à l'origine d'au moins 80 maladies
Gènes, hormones, microbiotes, environnement... Des maladies d'origine multifactorielle
Un contrôle efficace de certaines maladies grâce aux nouvelles biothérapies
Dossier réalisé en collaboration avec Olivier Boyer et Sophie Candon, unité 1234 Inserm/Université de Rouen Normandie, Physiopathologie, auto-immunité, maladies neuromusculaires et thérapie régénératrices, et Laboratoire d'immunologie et biothérapies, CHU de Rouen Normandie.
Alors qu’il est censé nous protéger contre les agents pathogènes (virus, bactéries…), notre système immunitaire peut parfois se déréguler. Il peut alors :
devenir trop sensible à certains constituants exogènes, et déclencher des allergies
ou bien
réagir contre des constituants du soi, et favoriser l’émergence de maladies auto-immunes
Les maladies auto-immunes forment un ensemble dans lequel on retrouve des maladies aussi différentes que le diabète de type 1, la sclérose en plaques, la polyarthrite rhumatoïde ou la maladie de Crohn. Elles correspondent toutes à des maladies chroniques déclenchées par la perte de tolérance immunologique de l’organisme face à ses propres constituants. Des effecteurs de l’immunité — anticorps ou cellules — engendrent alors des lésions cellulaires ou tissulaires responsables de symptômes plus ou moins sévères. Selon la nature de ces effecteurs, les lésions touchent un organe particulier (foie, pancréas, neurones...) ou différents tissus au sein de l’organisme.
Un certain degré d'autoréactivité du système immunitaire existe naturellement. Il permet aux lymphocytes B producteurs d’anticorps et aux lymphocytes T — cytotoxiques
ou producteurs de molécules favorisant l’inflammation (cytokines
) — de cibler nos cellules et leurs composants (ADN, protéines, noyaux cellulaires…). Mais au cours du développement, l’organisme développe une tolérance immunitaire vis-à-vis des constituants du soi. Ainsi, deux mécanismes de contrôle vont progressivement éduquer nos défenses et éviter qu’elles ne s’attaquent à nos tissus :
un mécanisme central qui permet d’éliminer les lymphocytes T fortement autoréactifs au niveau du thymus, et les lymphocytes B autoréactifs dans la moelle osseuse
un mécanisme périphérique qui, parallèlement, permet le contrôle des lymphocytes autoréactifs qui auraient échappé au mécanisme central
Plusieurs types de médiateurs assurent l’élimination ou le contrôle des effecteurs autoréactifs, comme les cytokines anti-inflammatoires ou les lymphocytes T régulateurs. Cependant, si ces processus sont imparfaits ou absents, l’autoréactivité est entretenue et conduit à l’apparition de la maladie auto-immune.
Les cellules dendritiques et les macrophages jouent également un rôle important dans le phénomène. Ces cellules assurent la présentation d’antigènes aux cellules de l’immunité, mais elles peuvent parfois enclencher une rupture de tolérance du soi en présentant des autoantigènes.
On estime aujourd’hui que 5 à 8% de la population mondiale est touchée par une maladie auto-immune. Si quelques-unes d’entre elles sont aussi fréquentes chez les hommes et les femmes (comme le diabète de type 1), 80% des cas concernent des femmes.
L’âge de survenue de ces maladies est très variable. Pour un certain nombre d’entre elles, l’âge auquel survient la maladie a cependant diminué au cours des dernières décennies : pour exemple, si le diabète de type 1 était une maladie de l’adolescent ou du jeune adulte, il n’est désormais pas rare de le diagnostiquer dès les premières années de vie. Par ailleurs, le nombre de maladies auto-immunes n’a cessé de croître depuis les années 1970 : on en recense actuellement environ 80.
Ces différentes dynamiques laissent penser qu’il existe une évolution de l’exposition à certains facteurs de risque. Reste à les identifier.
5 à 8 % de la population est concernée 8 personnes sur 10 sont des femmes
Facteurs favorisant la rupture du soi
Dans leur grande majorité, les maladies auto-immunes sont multifactorielles : cela rend difficile — pour ne pas dire impossible — d’en déterminer l’origine exacte. A quelques exceptions près, on estime qu’elle repose sur l’association de facteurs génétiques, endogènes, exogènes et/ou environnementaux :
Facteurs génétiques
La majorité des maladies auto-immunes est considérée comme dépendante d’une susceptibilité génétique, c’est-à-dire que leur développement est favorisé par une ou plusieurs particularités génétiques (ou polymorphismes). Parmi celles incriminées figurent d’abord des formes particulières des gènes HLA
, un ensemble de gènes qui codent pour des protéines permettant justement à l’organisme de reconnaître le soi du non-soi. Ces polymorphismes sont retrouvés parmi une part plus ou moins importante de patients atteints de spondylarthrite ankylosante (HLA-B27), polyarthrite rhumatoïde (HLA-DR4), diabète de type 1 (HLA-DR3/DR4) ou maladie cœliaque (HLA-DQ2)…
Des variations de la séquence de gènes non-HLA, peuvent également être impliquées. À mesure que la recherche progresse, leur nombre ne cesse de croître : ces variations affectent des gènes codant pour des médiateurs de la signalisation intracellulaire, pour les voies de costimulation, ou pour des facteurs de transcription
. Ainsi, des polymorphismes du gène PTPN22 ont été identifiés comme étant des facteurs de risque de polyarthrite rhumatoïde, de diabète de type 1 ou de lupus érythémateux disséminé (LED) ; ceux du gène IRF5 (impliqué dans la fonctionnalité des cytokines) ont été identifiés dans le LED, la polyarthrite rhumatoïde ou les sclérodermies systémiques ; ceux concernant le gène CD40 favoriserait la survenue de maladie de Basedow ou de polyarthrite rhumatoïde... Toutefois, ces variations génétiques ont un « poids » beaucoup moins important que les polymorphismes des gènes HLA dans le risque de survenue d’une maladie auto-immune.
Quelques très rares maladies auto-immunes ont une origine monogénique : dans ce cas, la mutation d’un seul gène est responsable de la pathologie, qui adopte alors le plus souvent une forme sévère. Ainsi, la mutation du gène AIRE, qui intervient normalement dans le contrôle central de l’auto-immunité au niveau du thymus, peut engendrer un syndrome polyendocrinien auto-immun. De la même façon, les mutations du gène FOXP3 réduisent le taux de cellules T régulatrices et favorisent l’apparition d’une entéropathie auto-immune de type 1 (syndrome IPEX). Les syndromes prolifératifs auto-immuns sont quant à eux liés à une anomalie de l’apoptose, engendrée par la mutation du gène FAS et, parfois du gène FASL.
Expression de AIRE (en vert) dans le thymus humain. La protéine AIRE (pour AutoImmune REgulator) joue un rôle clé dans l'éducation des lymphocytes. Or l'analyse moléculaire et cellulaire thymique a révélé qu'à partir de l'adolescence, les jeunes filles et les femmes ont moins d'AIRE que les hommes. Il en va de même chez les souris. A partir de la puberté, le taux élevé d'œstrogène chez les femmes inhibe l'expression d'AIRE dans le thymus, augmentant la susceptibilité aux maladies auto-immunes.
Facteurs endogènes
L’influence des hormones féminines sur les mécanismes de contrôle de l’auto-immunité pourrait expliquer pourquoi les femmes sont plus souvent affectées par ces maladies : le rôle des estrogènes et de la prolactine, sécrétée pour favoriser la lactation, a été confirmé par des travaux conduits dans différents modèles animaux.
D’autres facteurs, tels que l’inflammation chronique ou la libération d’autoantigènes séquestrés (c’est-à-dire non présentés au système immunitaire en condition normale), peuvent aussi avoir une influence sur le risque de développer une maladie auto-immune.
Enfin, les études expérimentales ou les données épidémiologiques décrivent clairement une association entre le microbiote intestinal, qui se situe à l’interface entre le système immunitaire et l’environnement, et la survenue d’une maladie auto-immune : la dysbiose, qui correspond à une modification qualitative et quantitative des différentes espèces colonisant notre système digestif par rapport aux conditions normales, est plus fréquente chez les malades que chez les personnes exemptes de maladies auto-immunes. La nature précise de cette dysbiose pourrait être différente, voire spécifique, selon la maladie considérée. Cependant, pour l’heure, il est difficile de savoir avec précision si la dysbiose est une cause ou une conséquence de la maladie. Le niveau de preuve actuel varie selon les pathologies : le rôle du microbiote est probable dans la maladie de Crohn, mais demande à être mieux décrit dans d’autres maladies auto-immunes (sclérose en plaques, lupus érythémateux disséminé, psoriasis…). Par ailleurs, les mécanismes à l’œuvre, expliquant comment les bactéries intestinales influencent l’immunité, restent à élucider.
Théorie de l’hygiène ou théorie du mimétisme ?
Notre susceptibilité à développer des maladies auto-immunes découlerait en partie de l’évolution récente des interactions entre immunité et exposition aux microorganismes viraux ou bactériens. Pour expliquer ce phénomène, deux théories non exclusives sont avancées :
La première correspond à la théorie hygiéniste qui suggère que la diminution de l’exposition naturelle aux agents infectieux (hygiène, alimentation…) et le recours croissant aux antibiotiques réduisent les capacités d’apprentissage et d’adaptation de l’immunité.
La seconde, dite du mimétisme moléculaire, repose sur l’idée que les antigènes présentés par les pathogènes peuvent présenter des similitudes structurelles avec les antigènes du soi, favorisant les réactions croisées lors d’un épisode infectieux : cela permettrait l’initiation puis le maintien d’une réaction immunitaire chronique.
Facteurs exogènes et environnementaux
L’exposition à certains composants ou certains pathogènes semble associée au risque de maladies auto-immunes, sans qu’un lien de causalité soit parfaitement établi. Ainsi, l’épidémiologie décrit une fréquence supérieure des infections préalables par les virus Epstein-Barr ou le cytomégalovirus chez les personnes atteintes. Le tabagisme, actif ou ancien, est aussi surreprésenté parmi les patients souffrant de polyarthrite rhumatoïde, de sclérose en plaques, de dysthyroïdie auto-immune… Certains polluants environnementaux, les ultraviolets, le stress ou la nutrition sont aussi suspectés, mais leur rôle reste à démontrer.
Récemment il a été démontré qu’une famille de médicaments anticancéreux modulant l’immunité constitue un facteur de risque potentiel d’auto-immunité : les inhibiteurs des points de contrôle immunitaires (atézolizumab, ipilimumab, nivolumab, pembrolizumab). Ces molécules ont été développées pour intensifier les défenses de l’organisme contre les cellules tumorales, mais les personnes qui ont été soignées par ces médicaments développent plus fréquemment des maladies auto-immunes (diabète, vitiligo, thyroïdite...). Il est maintenant nécessaire de déterminer s’ils déclenchent le processus d’auto-immunité ou s’ils ne font que favoriser son développement chez des sujets initialement prédisposés.
Rouages De l’auto-immunité aux symptômes...
Les deux familles d’effecteurs impliqués dans l’auto-immunité – anticorps et lymphocytes T – engendrent des lésions tissulaires qui leurs sont spécifiques.
Les autoanticorps
Les autoanticorps sont souvent les effecteurs dont l’impact lésionnel est prépondérant. En effet, ils favorisent la mort de la cellule cible (ex : anémies hémolytiques) et forment des complexes dits immuns avec l’antigène, provoquant des lésions vasculaires ou rénales (ex : néphropathie glomérulaire, lupus érythémateux disséminé). En se fixant sur des structures membranaires extracellulaires (récepteurs, antigènes…), ces anticorps peuvent aussi perturber le mécanisme biologique normal auquel ces cellules participent habituellement (ex : dysthyroïdie). Ces effecteurs peuvent, dans certains cas, être transmis de la mère à l’enfant par voie placentaire (ex : anticorps ciblant l’acétylcholine dans la myasthénie).
Les lymphocytes T autoréactifs
Les lymphocytes T autoréactifs jouent également un rôle significatif, en favorisant la lyse des cellules cibles, directement par cytotoxicité, ou indirectement par la production de cytokines. Pour exemple, ces cellules sont principalement responsables de la destruction des cellules bêta des îlots de Langerhans dans le diabète de type 1, ou de la destruction des gaines de myélinedans la sclérose en plaques.
Les médiateurs de l’inflammation
Enfin, la composante inflammatoire, quasi-systématique en cas de maladie auto-immune, joue un rôle important : souvent asymptomatique au début de la maladie, elle a tendance à se chroniciser et devenir cliniquement significative (rougeur, gonflement, douleur…). Petit à petit, l’inflammation favorise des modifications locales de l’organisation cellulaire et tissulaire (granulome inflammatoire, destruction et réparation tissulaires, fibrose…) qui peuvent devenir difficiles à normaliser. Cette inflammation est notamment médiée par les cytokines, qui sont des petites molécules circulantes de différente nature (TNF alpha, interleukines…).
Lupus érythémateux disséminé (LED). Glomérule rénal atteint de lupus érythémateux disséminé (LED), maladie auto-immune chronique.
...et des symptômes au diagnostic
Le diagnostic d’une maladie auto-immune repose sur des éléments cliniques et biologiques, parfois complétés de données génétiques et d’imagerie.
Des symptômes ou signes cliniques font généralement suspecter un diagnostic particulier et incitent le médecin à réaliser ou prescrire des examens. Beaucoup de maladies auto-immunes se manifestent par des phases de poussées, durant lesquelles les symptômes s’intensifient, entrecoupées par des périodes de rémission. Mais d’autres sont associées à des symptômes constants, pouvant évoluer avec le temps. Outre l’examen clinique, une imagerie médicale peut être nécessaire pour observer les lésions des organes touchés.
Si la piste semble solide, des examens biologiques spécifiques sont prescrits : ils permettent de rechercher des biomarqueurs
propres à l’auto-immunité, comme des complexes immuns circulants, des autoanticorps spécifiques (anticorps anti-insuline, anti-ADN, APL, ANCA, CCP, facteur rhumatoïde... selon la pathologie) . La recherche de biomarqueurs propres à l’inflammation est également conduite, notamment en évaluant la vitesse de sédimentation, le taux de protéine-C réactive (CRP) ou la présence de fractions du complément.
Lorsqu’un gène de susceptibilité est connu comme étant fortement corrélé au risque de développer la maladie suspectée, un test génétique complète le diagnostic. Le gène HLA B27 est par exemple recherché dans le cadre du diagnostic de spondyloarthrite.
Des traitements de plus en plus spécifiques
Contrôler les symptômes (antalgiques, anti-inflamatoires, hormones...). Contrôler l'auto-immunité (immunosuppresseurs, biothérapies). Eliminer ou neutraliser les autoanticorps (plasmaphérèse, immunoglobuline intraveineuse)
Chaque maladie auto-immune répond à une prise en charge spécifique. Des traitements permettent de contrôler les symptômes de la maladie : antalgiques contre la douleur, anti-inflammatoires contre la gêne fonctionnelle articulaire, médicaments substitutifs permettant de normaliser les troubles endocriniens (insuline dans le diabète, thyroxine dans l’hypothyroïdie…), etc…
Des médicaments permettant de contrôler ou d’inhiber l’auto-immunité offrent aussi un moyen de limiter les symptômes et la progression des lésions tissulaires. Ils doivent généralement être pris de façon chronique car ils ne permettent pas de guérir la maladie. De plus, ils ne sont pas spécifiques des cellules effectrices de l’auto-immunité et interfèrent avec certaines fonctions générales du système immunitaire. Historiquement, les médicaments immunosuppresseurs (corticoïdes, cyclophosphamide, méthotrexate, azathioprine, ciclosporine…) sont utilisés car ils interagissent sur des effecteurs centraux du système immunitaire et permettent de limiter son activité de façon globale. Ils sont souvent associés à un risque d’infection accru et nécessitent en conséquence un suivi régulier. Depuis une petite vingtaine d’années, grâce aux progrès des biotechnologies, des biothérapies sont développées : elles offrent une meilleure maîtrise des symptômes et des risques de lésions. Contrairement aux immunosuppresseurs, ce sont des molécules qui ciblent spécifiquement un des acteurs clés impliqués dans le processus pathologique concerné. Une biothérapie est généralement spécifique d’une maladie auto-immune, ou de plusieurs lorsqu’elles partagent des effecteurs communs. Ils sont généralement utilisés lorsque la maladie est sévère ou qu’elle ne répond pas, ou insuffisamment, aux immunosuppresseurs.
Les anti-TNF alpha font partie des premières biothérapies développées dans le traitement des maladies auto-immunes. Ils inhibent les mécanismes inflammatoires que le TNF alpha ou son récepteur déclenche habituellement. Plusieurs sont aujourd’hui commercialisés (infliximab, adalimumab, certolizumab pegol, étanercept...) dans le traitement de la polyarthrite rhumatoïde, la maladie de Crohn ou les spondyloarthrites… D’autres molécules ont été développés depuis, ciblant l’IL-1 (canakinumab, anakinra), l’IL-6 (tocilizumab), l’IL-12/IL-23 (ustékinumab), la fraction C5 du complément (éculizumab) ou les récepteurs à la sphingosine 1-phosphate (fingolimod)… Certains ont une efficacité élevée et permettent de contrôler totalement les symptômes de la maladie.
Ces traitements sont pris de façon chronique. Cependant, des essais cliniques conduits dans la maladie de Crohn ou la polyarthrite rhumatoïde suggèrent que des patients ayant bénéficié d’une rémission durable des symptômes durant plusieurs années pourraient arrêter le traitement sans réactivation de la maladie. Des études complémentaires sont évidemment nécessaires pour confirmer ces premiers résultats et évaluer si cette rémission est durable.
De façon plus spécifique, d’autres traitements sont parfois envisagés dans quelques pathologies très spécifiques : la plasmaphérèse permet ainsi l’élimination des auto-anticorps par filtration du sang qui est ensuite réinjecté au patient. Cette stratégie est utilisée dans le traitement de la myasthénie ou du syndrome de Guillain-Barré. Des immunoglobulines intraveineuses (IgIV), constituées à partir d’immunoglobulines issues de dons du sang, sont aussi utilisées pour neutraliser ou moduler les anticorps pathogènes ou pour réguler la production d’autoanticorps dans le purpura thrombocytémique idiopathique
(PTI), le syndrome de Guillain-Barré ou la maladie de Kawasaki.
Les enjeux de la recherche
Identifier les facteurs de risque émergents
L’augmentation de la fréquence des maladies auto-immunes dans les pays occidentaux au cours des dernières décennies laissent penser que l’exposition ou l’influence de certains facteurs de risque a évolué.
Les études dédiées au microbiote sont particulièrement nombreuses car l’influence des microorganismes qui colonisent notre système digestif semble déterminante.
Les facteurs génétiques continuent à être explorés : facteurs monogéniques rares, gènes de susceptibilité… La prépondérance féminine liée à l’auto-immunité pourrait ainsi reposer sur une origine génétique. Des gènes favorisant l’auto-immunité portés par le chromosome X pourraient échapper à des mécanismes d’inactivation qui en contrôle normalement l’expression, ce qui expliquerait la prévalence
supérieure des maladies auto-immunes chez les femmes. C’est notamment le cas pour le gène TLR7, qui semble être un facteur de risque de lupus érythémateux systémique (LED) et probablement d’autres pathologies auto-immunes.
Outre les mutations génétiques, des variations épigénétiques — qui modifient de manière pérenne la façon dont nos gènes s’expriment — font aussi l’objet de recherches. Ces variations seraient engendrées par l’influence de facteurs environnementaux, de l’hygiène de vie ou de l’exposition à certains traitements.
Identifier de nouveaux médiateurs pour développer de nouveaux traitements
L’objectif ultime de la recherche clinique est de pouvoir supprimer spécifiquement le mécanisme d’origine d’une maladie auto-immune, sans modifier le fonctionnement normal du reste du système immunitaire. Il reste difficile à atteindre car les cascades biologiques normales et pathologiques présentent souvent des effecteurs communs. Pour autant, la recherche progresse en ce sens grâce aux progrès importants réalisés en recherche fondamentale, préalable indispensable au développement des thérapies ciblées.
Les chercheurs sont aidés par la disponibilité de modèles animaux mimant les pathologies auto-immunes humaines. L’analyse « en cellule unique » offre également de nouvelles perspectives pour mieux appréhender la diversité de certains types cellulaires impliqués dans l’auto-immunité : en caractérisant les interactions cellulaires au niveau d’une seule cellule, elle permet de distinguer les spécificités et caractéristiques de chaque cellule d’intérêt, plutôt que de moyenner les comportements cellulaires comme le font les méthodes d’études conventionnelles (en pool cellulaire).
Grâce à ces efforts de recherche, les traitements ciblés se multiplient, notamment dans le traitement des maladies inflammatoires rhumatismales ou gastro-intestinales. Elles bénéficient désormais aussi à des pathologies réputées plus difficiles à traiter comme la sclérose en plaques (fingolimod, natalizumab).
Limiter les risques de complications, notamment infectieuses, sous traitement modulant l’immunité reste une gageure. Le natalizumab indiqué dans la sclérose en plaques en est une bonne illustration : ce médicament cible un récepteur (intégrine
VLA-4) permettant aux lymphocytes T autoréactifs de passer la barrière hémato-encéphalique pour détruire la gaine de myéline indispensable au bon fonctionnement des neurones. Il accroît parallèlement le risque de leucoencéphalopathie multifocale progressive (LEMP), une complication très rare mais sévère, liée à la réactivation du virus JC préalablement présent, à l’état dormant, dans le système nerveux. D’autres approches, visant à favoriser préférentiellement l’action des lymphocytes T régulateurs pourraient, en conséquence être plus efficace. Des expérimentations utilisant des injections d’interleukine-2 (IL-2, voir plus loin) sont en cours d’évaluation.
Vers un traitement par thérapie cellulaire ?
Les cellules souches mésenchymateuses (multipotent mesenchymal stromal cells) ont la capacité de se multiplier indéfiniment et de pouvoir se différencier en de nombreux type de cellules. Ces cellules souches sont faciles à prélever, à cultiver et à différencier dans le type de cellule souhaité (en adaptant leur milieu de culture) in vitro. De plus, elles ne sont pas ou peu reconnues par les cellules immunitaires effectrices. La greffe autogénique et allogénique est donc plus facilement envisageable à partir de ces cellules qu’à partir de cellules souches hématopoïétiques.
Les cellules souches mésenchymateuses permettraient de régénérer un tissu pathologique comme, par exemple, le tissu articulaire abîmé par les mécanismes auto-immuns et inflammatoires locaux. Elles permettraient également de favoriser un phénomène d’immunomodulation, grâce à l'activité immunorégulatrice qui leur est propre. Ainsi, elles font l’objet de nombreux travaux de recherche dans les pathologies inflammatoires rhumatismales et gastro-intestinales. Le programme européen ADIPOA teste actuellement leur apport dans l'arthrose par injection de cellules issues du tissu adipeux
L’autogreffe de cellules souches hématopoïétiques (CSH) reste néanmoins une option intéressante pour certaines pathologies. Des études cliniques, notamment conduites en France, ont par exemple permis de les greffer avec succès chez des sujets souffrant de sclérodermie, après prélèvement initial, chimiothérapie forte dose et radiothérapie corporelle, puis réinjection des CSH.
De nouveaux effecteurs du contrôle immunitaire
L’identification des effecteurs de l’auto-immunité permet de disposer de biomarqueurs favorisant à la fois le diagnostic, le développement de nouveaux traitements et le suivi de l’efficacité thérapeutique.
Les autoanticorps peuvent revêtir un rôle crucial dans ce cadre, chacun d’entre eux pouvant offrir une aide diagnostique spécifique d’un phénotype de maladie. Ainsi, chacun des phénotypes constituant la famille des dermatomyosites semble associé à un type d’autoanticorps particulier. Chacun d’eux pourrait aussi constituer la cible d’un nouveau traitement. Mais la recherche est plus dense concernant les autres médiateurs de l’immunité ou de l’auto-immunité.
Ainsi, dans la grande famille des interleukines, molécules médiatrices des fonctions immunitaires, l’interleukine-2 (IL-2) suscite un nouvel espoir dans la lutte contre les maladies auto-immunes. Connue pour jouer un rôle déterminant dans la réponse aux infections et aux processus tumoraux, l’IL-2 intervient aussi sur la différenciation des lymphocytes T régulateurs et celle des lymphocytes T mémoire. Ces derniers, comme leur nom l’indique, assurent la mémoire de nos défenses en cas de nouvelle exposition à un antigène déjà rencontré. Ainsi, l’IL-2 aurait un rôle actif sur la tolérance immunitaire face aux auto-antigènes. La compréhension de ses fonctions a permis de développer de nouvelles approches thérapeutiques pour combattre les maladies auto-immunes : l’intérêt de l’IL-2 fait l’objet d’études cliniques dans le traitement de la sclérose en plaques, du diabète de type 1, de la sclérodermie ou du syndrome de Sjögren.
Image réalisée en microspopie par fluorescence
Îlot pancréatique infiltré par des Treg (en rose). Les lymphocytes T régulateurs CD4+CD25+Foxp3+ jouent un rôle majeur dans le contrôle du diabète de type 1, en inhibant les cellules capables de détruire les cellules pancréatiques productrices d’insuline. Chez des souris récemment diabétiques, l’administration de faibles doses d’IL-2 entraîne une rémission de la maladie après seulement 5 jours de traitement chez 60 % des animaux. Un essai clinique devrait être organisé pour étudier le potentiel thérapeutique de cette cytokine dans le diabète humain.
Parallèlement, si les lymphocytes T jouent un rôle prépondérant dans l’auto-immunité, il semble de plus en plus évident que les lymphocytes B jouent un rôle déterminant sur la régulation d’autres effecteurs : celle des lymphocytes T, via la production d’IL-10, et celle des cellules dendritiques qui présentent les antigènes. Les lymphocytes B suscitent donc un regain d’intérêt, parallèlement aux molécules qui les influencent directement, comme BAFF (B cell activating factor), une cytokine qui joue un rôle important pour leur survie.
Enfin, si la plupart des traitements ciblés existant cherchent à contrôler les cascades réactionnelles impliquées dans des interactions entre cellules, d’autres commencent à être développés pour contrer les voies de signalisation intracellulaires activées dans les lymphocytes effecteurs de l’auto-immunité. Parmi elles, la voie de signalisation JAK/STAT est particulièrement intéressante. Elle est étudiée dans la polyarthrite rhumatoïde car elle contrôle la régulation de gènes codant pour des cytokines pro-inflammatoires. Deux inhibiteurs de JAK2 (tofacitinib et baricitinib) font notamment l’objet d’études cliniques actuellement. Ils pourraient constituer les premières molécules ciblant les processus intracellulaires liés à l’auto-immunité, avant d’autres voies pour lesquelles les travaux sont moins avancés (MAP kinases, PI3 kinases).
Consortium européen PRECISADS : vers une nouvelle classification
Les maladies auto-immunes systémiques (Systemic Autoimmune DiseaseS ou SADS) forment un ensemble de maladies chroniques aux manifestations cliniques diverses. Une même maladie peut aussi se manifester par des symptômes plus ou moins spécifiques, offrant une certaine hétérogénéité à chacune de ces entités.
Pour les scientifiques, cette hétérogénéité s’explique par la classification actuelle, portant essentiellement sur des critères cliniques : selon eux, il est probable que des techniques innovantes de type « omiques » (génomique, transcriptomique, épigénomique, métabolomique, protéomique) puissent apporter une nouvelle classification, reposant sur des anomalies et mécanismes physiopathologiques biologiques communs, pour, in fine, aboutir à de nouveaux traitements, plus spécifiques.
En ce sens, un consortium de recherche, PRECISADS, a été lancé entre 9 pays européens dont la France (avec CHRU de Brest / unité Inserm 1227). Avec le recrutement de près de 3 000 patients, ce projet européen vise notamment à identifier des signatures moléculaires spécifiques qui permettront de mieux classifier, puis traiter les patients. PRECISESADS devrait permettre d’apporter un nouvel éclairage sur l’étiologie des maladies auto-immunes.
Etude géographique du birdshot par Horus birdshot base 2017

Mes ami(e)s,
Je vous prie de bien vouloir trouver ci-joint, une étude exhaustive sur les visites du site www.horus-birdshot.fr, site dédié à la rétinochoroidite de type birdshot, et ce, dans le monde entier.
Ce dossier est en français, mais il contient essentiellement des chiffres, qui ne nécessitent absolument aucune traduction linguistique.
En tant que malade, mais néanmoins scientifique, j’ai souhaité que cette étude soit la plus « honnête » possible, et vous ne trouverez donc pas de supputations aléatoires, ni de tentatives, vaines, d’estimations aléatoires de l’étendue de la maladie.
Néanmoins, et selon les règles que je me suis fixées et que je vous énoncerai, il est « raisonnablement » possible de faire un point « raisonnable » sur l’étendue géographique de la maladie et de son ampleur selon les régions du monde.
J’espère que vous apprécierez ce travail étayé par 7 ans de travail en amont sur le site wwwhorus-birdshot.fr.
1/ les règles de travail :
Les chiffres présents sur cette étude sont basés sur les éléments simples suivants :
1. Recherche Google sur la « maladie du birdshot », ou « birdshot disease », ou « rétinochoroidite de type birdshot » ou « birdshot retinopathy ».
2. Ces contacts ont eu lieu du 5 janvier 2017 au 5 janvier 2018
3. Les contacts sont restés plus de 30 secondes sur le site.
On peut donc, à priori, estimer que ces contacts sont de bonne qualité pour qualifier une étude sur la maladie (à l’erreur près)
2/ Relativité :
1. Nous sommes surtout dépendant du fait que nos docteurs ne sont pas formés sur la maladie (pour une grande partie) et si cela est de moins en moins vrai en France, c’est une certitude sur l’ensemble de la planète. Aussi, si l’on estime à je cour, environ 1000 à 1500 malades dans le monde (déclarés), on ne peut pas connaitre de manière certaine, les personnes atteintes de la maladie.
2. Bien que le site soit international, la majorité de commentaires sont en français (avec un onglet en anglais néanmoins) ce qui ne facilite pas forcement la communication à l’international.
3. Une partie non négligeable du monde n’a pas nécessairement accès à l’internet et ne peut donc pas aisément se renseigner. Nous perdons donc une bonne partie des informations.
Aussi, malgré ces inconvénients, et selon la loi des grands nombres, il est à priori « raisonnable » de penser, qu’au moins géographiquement, les informations figurant sur cette étude sont cohérentes.
Comme vous pourrez le constater que n’y ai ajouté aucune conjecture d’aucune sorte, afin que tout un chacun puisse se faire sa propre opinion sur l’étendue de la maladie.
Pour notre part, nous travaillons essentiellement avec le Pr Brézin, de l’APHP à Paris, ainsi qu’avance d’autres hôpitaux en France, car, à ce jour, nous représentons le plus de cas avérés (plus de 500 en France). Le Pr Brézin est le spécialiste mondial de cette maladie et l’ensemble de vos médecins et professeurs le connaissent, connaissent son travail et le respectent.
Bien entendu, je reste à votre disposition pour tout renseignement complémentaire, toute suggestion, toute remarque.
Sylvain MIGNON
Président créateur de l’association Horus birdshot
Association loi 1901 dédiée à la rétinochoroidite de type birdshot.
Mes ami(e)s,
Je vous prie de bien vouloir trouver ci-joint, une étude exhaustive sur les visites du site www.horus-birdshot.fr, site dédié à la rétinochoroidite de type birdshot, et ce, dans le monde entier.
Ce dossier est en français, mais il contient essentiellement des chiffres, qui ne nécessitent absolument aucune traduction linguistique.
En tant que malade, mais néanmoins scientifique, j’ai souhaité que cette étude soit la plus « honnête » possible, et vous ne trouverez donc pas de supputations aléatoires, ni de tentatives, vaines, d’estimations aléatoires de l’étendue de la maladie.
Néanmoins, et selon les règles que je me suis fixées et que je vous énoncerai, il est « raisonnablement » possible de faire un point « raisonnable » sur l’étendue géographique de la maladie et de son ampleur selon les régions du monde.
J’espère que vous apprécierez ce travail étayé par 7 ans de travail en amont sur le site wwwhorus-birdshot.fr.
1/ les règles de travail :
Les chiffres présents sur cette étude sont basés sur les éléments simples suivants :
1. Recherche Google sur la « maladie du birdshot », ou « birdshot disease », ou « rétinochoroidite de type birdshot » ou « birdshot retinopathy ».
2. Ces contacts ont eu lieu du 5 janvier 2017 au 5 janvier 2018
3. Les contacts sont restés plus de 30 secondes sur le site.
On peut donc, à priori, estimer que ces contacts sont de bonne qualité pour qualifier une étude sur la maladie (à l’erreur près)
2/ Relativité :
1. Nous sommes surtout dépendant du fait que nos docteurs ne sont pas formés sur la maladie (pour une grande partie) et si cela est de moins en moins vrai en France, c’est une certitude sur l’ensemble de la planète. Aussi, si l’on estime à je cour, environ 1000 à 1500 malades dans le monde (déclarés), on ne peut pas connaitre de manière certaine, les personnes atteintes de la maladie.
2. Bien que le site soit international, la majorité de commentaires sont en français (avec un onglet en anglais néanmoins) ce qui ne facilite pas forcement la communication à l’international.
3. Une partie non négligeable du monde n’a pas nécessairement accès à l’internet et ne peut donc pas aisément se renseigner. Nous perdons donc une bonne partie des informations.
Aussi, malgré ces inconvénients, et selon la loi des grands nombres, il est à priori « raisonnable » de penser, qu’au moins géographiquement, les informations figurant sur cette étude sont cohérentes.
Comme vous pourrez le constater que n’y ai ajouté aucune conjecture d’aucune sorte, afin que tout un chacun puisse se faire sa propre opinion sur l’étendue de la maladie.
Pour notre part, nous travaillons essentiellement avec le Pr Brézin, de l’APHP à Paris, ainsi qu’avance d’autres hôpitaux en France, car, à ce jour, nous représentons le plus de cas avérés (plus de 500 en France). Le Pr Brézin est le spécialiste mondial de cette maladie et l’ensemble de vos médecins et professeurs le connaissent, connaissent son travail et le respectent.
Bien entendu, je reste à votre disposition pour tout renseignement complémentaire, toute suggestion, toute remarque.
Sylvain MIGNON
Président créateur de l’association Horus birdshot
Association loi 1901 dédiée à la rétinochoroidite de type birdshot.
maladie auto immune
Maladie auto-immune
Un article de Wikipédia, l'encyclopédie libre.
Aller à : Navigation, rechercher
Les maladies auto-immunes sont dues à une hyperactivité du système immunitaire à l'encontre de substances ou de tissus qui sont normalement présents dans l'organisme.
Parmi ces maladies peuvent être cités la sclérose en plaques, le diabète de type 1 (jadis appelé " diabète juvénile " ou " diabète insulino-dépendant "), le lupus, les thyroïdites auto-immunes, la polyarthrite rhumatoïde, la spondylarthrite ankylosante, le syndrome de Goujerot-Sjögren, la maladie de Crohn, etc.
Leur cause n'est pas encore bien élucidée : certaines d'entre elles sont considérées comme des maladies d'abondance. Le lien est par exemple avéré entre l'arthrite et l'obésité, et l'OMS déclare que l'arthrite est plus fréquente dans les pays développés. La plupart des maladies auto-immunes sont probablement le résultat de causes multiples, telles qu'une prédisposition génétique stimulée par une infection, associée à la présence d'une substance chimique ou aliment.
Les femmes semblent être plus touchées par les maladies auto-immunes. On ignore pourquoi, bien qu'il ait été prouvé que les taux d'hormones soient liés à la gravité de certaines maladies auto-immunes dont la sclérose en plaques1.
antigen hla a 29
HLA-A29
From Wikipedia, the free encyclopedia
Jump to: navigation, search
HLA-A29
(MHC Class I, A cell surface antigen)
HLA-A29
Protein
transmembrane receptor/ligand
Structure
?? heterodimer
Subunits
HLA-A*29--, ?2-microglobulin
Older names "A19"
subtype allele Available structures
A29.1 *2901
A29.2 *2902
Alleles link-out to IMGT/HLA database at EBI
HLA-A29 (A29) is a human leukocyte antigen serotype within HLA-A serotype group. The serotype is determined by the antibody recognition of ?29 subset of HLA-A ?-chains. For A29, the alpha "A" chain are encoded by the HLA-A*29 allele group and the ?-chain are encoded by B2M locus.[1] This group currently is dominated by A*2902. A29 and A*29 are almost synonymous in meaning. A29 is a split antigen of the broad antigen serotype A19. A29 is a sister serotype of A30, A31, A32, A33, and A74.
Further information: HLA serotypes explained
A29 is more common in Western Africa and Southwestern Europe.
HORUS BIRDSHOT
association loi 1901 à but non lucratif
17 rue du Ferlin
85670 FALLERON
tel 03.26.02.17.93 -
GSM 06.24.91.49.31
email : contact@horus-birdshot.fr
ACCES FACEBOOK
une maladie atteignant les personnes de type "caucasien"
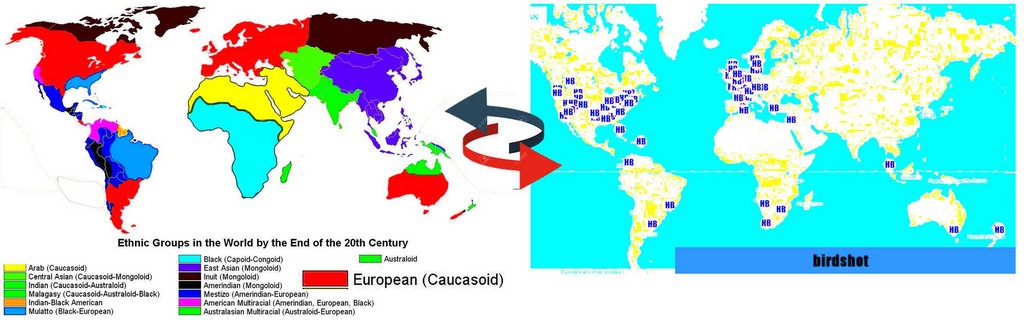
Quand on parle de maladie atteignant les population de type 'caucasien". Une bonne image vaut tous les discours.
Vous trouvez ci-joint, le parallèle entre les zones (en rouge) des populations de type caucasien et la cate des hopitaux traitant des malades du birdshot.
1949 decouverte

première description de la maladie en 1949.